
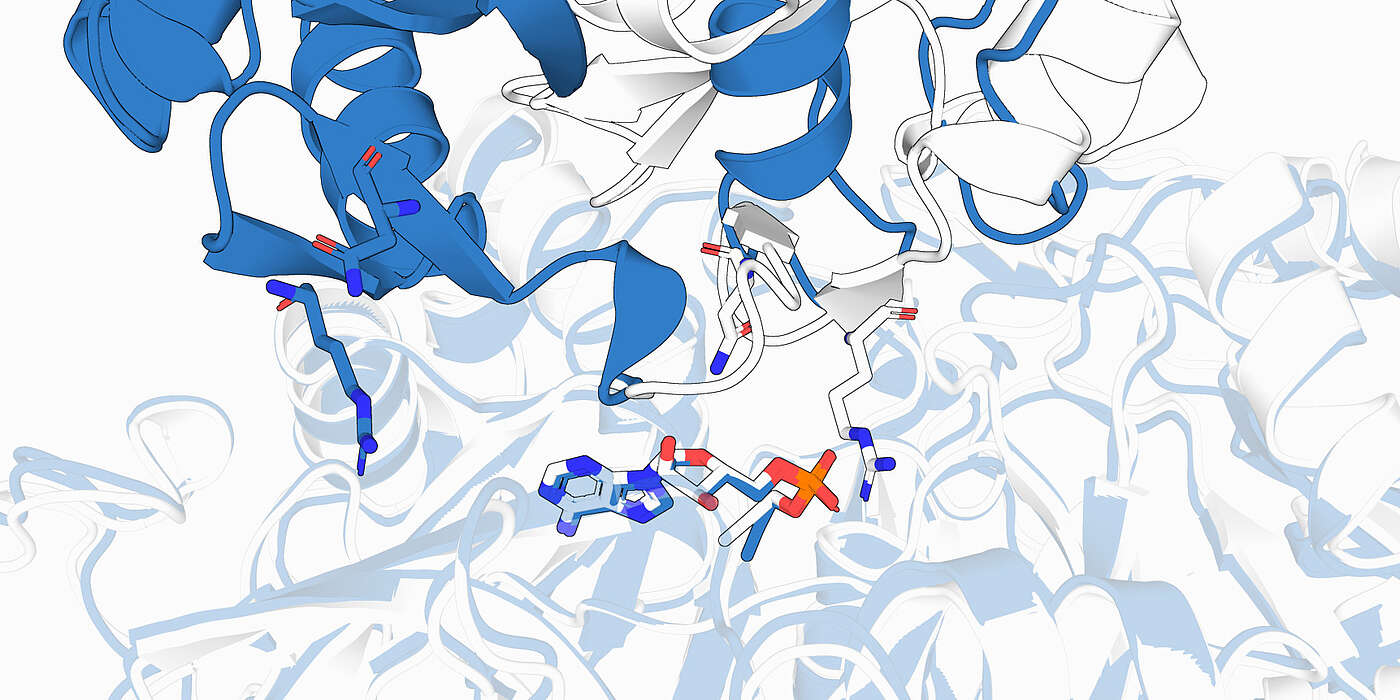
Als Google DeepMind vor wenigen Jahren mit AlphaFold die dreidimensionale Struktur von Proteinen nahezu perfekt vorhersagte, weckte dies grosse Hoffnungen: Könnte Künstliche Intelligenz (KI) bald auch vorhersagen, welche Moleküle wo und wie an ein Protein binden und dessen Funktion beeinflussen? Genau diese Informationen braucht es, um neue Medikamente zu entwickeln. Der Einsatz von KI könnte die Wirkstoffentwicklung daher enorm beschleunigen.
In ihrer kürzlich in «Nature Structural & Molecular Biology» publizierten Arbeit dämpfen Dr. Janani Durairaj und Prof. Torsten Schwede vom Biozentrum, Universität Basel, die Erwartung und Euphorie ums KI-gestützte Wirkstoffdesign. «Bei Molekülen mit bekanntem Muster können heutige KI-Modelle zwar zuverlässige Vorhersagen zur Bindung an ein Protein treffen», so Durairaj. «Doch bei neuartigen Verbindungen tun sie sich noch schwer.» In der Arzneimittelforschung geht es jedoch selten darum, Altbekanntes zu reproduzieren, sondern um Innovation.
KI-Modelle auf dem Prüfstand
Bislang war unklar, ob die heutigen KI-Modelle tatsächlich lernen, wie Moleküle – sogenannte Liganden – an Proteine binden oder ob sie lediglich Beispiele aus ihren Trainingsdaten «wiedererkennen». Um dies zu überprüfen, haben die Forschenden einen neuen, umfangreichen Testdatensatz Runs N’Poses zusammengestellt. Dieser enthält detaillierte Informationen von über 2600 Protein-Liganden-Komplexen, die experimentell bestimmt und erst nach dem Trainingsende der gängigen KI-Modelle veröffentlicht wurden. Anschliessend untersuchten die Forschenden, wie gut vier der modernsten KI-Systeme mit unbekannten Molekülen zurechtkommen.
«Die Bilanz war schlichtweg ernüchternd. Die Modelle schneiden gut ab, wenn ein Ligand und seine Wechselwirkung mit Proteinen bereits Gelerntem ähnelt, die chemische Struktur der KI also aus den Trainingsbeispielen vertraut ist», sagt Durairaj. «Doch bei unbekannten Interaktionen, die die KI zuvor noch nie gesehen hat, nimmt die Qualität der Vorhersage stark ab.» Die KI «erinnert» sich an Bekanntes aus den Trainingsdatensätzen und reproduziert dies bei den Vorhersagen. Die zugrundeliegenden biophysikalischen Prinzipien, wie Protein und Ligand miteinander interagieren, versteht sie jedoch nicht.
Mit ihrem frei verfügbaren Trainingsdatensatz Runs N’ Poses haben die Forschenden eine Grundlage geschaffen, mit der künftige KI-Modelle realistischer evaluiert und trainiert werden können. «Beim Wirkstoffdesign hat die KI ein riesiges Potenzial», sagt Schwede. «Aber wir müssen genau hinschauen, wo sie heute steht, was sie kann und was noch nicht. Nur dann können wir KI-Modelle dahingehend trainieren, dass wir mit ihrer Hilfe tatsächlich neue Wirkstoffe designen können.»
Bewertung von KI-Modellen zu optimistisch
Die Studie zeigt, dass frühere Erfolgsmeldungen oftmals zu optimistisch waren. Viele Tests prüfen die KI mit Aufgaben, die den Trainingsbeispielen stark ähneln. Das ist, als würde man jemanden für eine Prüfung loben, obwohl die Fragen identisch zu den Übungsaufgaben waren. Die Forschenden plädieren daher für vielfältigere Trainingsdaten und realistischere Tests. Gerade bei der Entwicklung von Arzneistoffen braucht es Innovation, weil bisherige Medikamente beispielsweise nicht mehr helfen, etwa bei resistenten Keimen oder Krebs, oder weil es einfach noch keine Therapie gibt, wie bei einigen seltenen Erkrankungen.
Bewertungsmethoden für KI-Vorhersagen
In einer zweiten Studie zeigt das Team um Torsten Schwede zudem auf, warum es neue Bewertungsverfahren für KI-Vorhersagen braucht. Moderne KIs generieren Millionen von Vorhersagen für immer grössere und komplexere Proteinstrukturen und Interaktionen mit Liganden.
Gleichzeitig werden die zugrundeliegenden Datensätze umfangreicher und die experimentell bestimmten Vergleichsstrukturen grösser. Zu bewerten, wie zuverlässig die Vorhersagen sind, wird daher immer schwieriger. In «Nature Methods» gehen die Forschenden auf die Schwächen bisheriger Verfahren ein und stellen neue Ansätze vor, um KI-Vorhersagen künftig verlässlicher einordnen zu können.
Publikationen:
Peter Škrinjar, Jérôme Eberhardt, Gabriel Studer, Gerardo Tauriello, Torsten Schwede, Janani Durairaj. Evaluating generalization in protein-ligand cofolding methods. Nature Structural & Molecular Biology, published online 8 May 2026
Gabriel Studer, Xavier Robin, Stefan Bienert, Janani Durairaj, Peter Škrinjar, Gerardo Tauriello, Andrew Waterhouse, Torsten Schwede. Comparing macromolecular complexes - a fully automated benchmarking suite. Nature Methods; published online 22 December 2025
Kontakt: Kommunikation, Katrin Bühler