
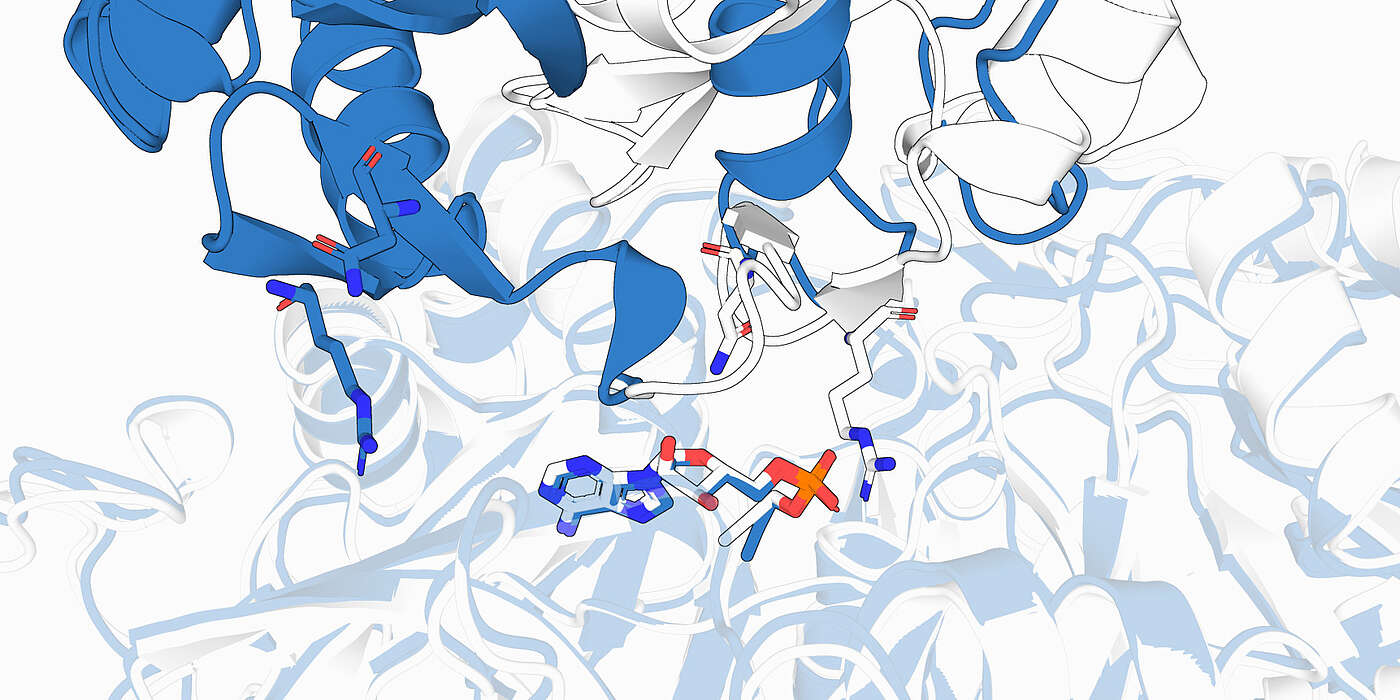
When Google DeepMind’s AlphaFold system stunned the scientific community by predicting the three-dimensional structure of proteins with near-perfect accuracy, it raised high hopes: Could artificial intelligence (AI) soon also predict which small molecules bind to a protein and how these interactions might influence protein function? Such information is essential for designing new drugs, and therefore the use of AI could greatly accelerate drug discovery.
In a recent study published in “Nature Structural & Molecular Biology,” Dr. Janani Durairaj and Prof. Torsten Schwede from the Biozentrum, University of Basel, temper expectations and euphoria around AI-assisted drug design. “For molecules with familiar patterns, today’s AI models can indeed make reliable predictions about how they interact with proteins,” says Durairaj. “But they still struggle with entirely new molecules or even known molecules binding in new ways.” This limitation poses a major challenge for drug discovery, where the goal is rarely to reproduce what is already known, but rather to innovate.
AI models put to the test
Until now, it has been unclear whether modern AI models learn how small molecules – known as ligands – interact with proteins, or whether they merely “recognize” and reproduce examples from their training data. To evaluate this, the researchers introduced a new, large-scale benchmark dataset called Runs N’ Poses. The dataset contains detailed structural information on more than 2,600 high-resolution protein–ligand complexes that were determined experimentally and released after the training cutoff of current AI systems. This allowed the team to assess how well four of the leading AI systems cope with unknown molecules.
“The results were sobering,” says Durairaj. “The models succeed when faced with ligands similar to what has been learned, in other words, when their chemical structure and interaction is familiar to the AI from the training data. But for truly unknown binding modes that the AI has never seen before, prediction quality decreases significantly.” The current AI systems rely on memorizing and interpolating between known examples from their training data rather than understanding the underlying biophysical principles that govern protein–ligand interactions.
With their open-source Runs N’ Poses benchmark dataset, the researchers provide a more realistic basis for evaluating and training future AI models. “AI has enormous potential for drug development,” says Schwede. “But we need to be honest about what the technology can and cannot yet achieve. Only with realistic benchmarks can we train AI systems that genuinely help us discover new drugs.”
AI models rated too optimistically
The study also shows that earlier evaluations of AI-driven drug discovery were often overly optimistic. Many benchmarks test AI models on problems that closely resemble their training data. That’s like praising someone for passing an exam when the questions are identical to the practice exercises. The researchers therefore advocate for more diverse training data and more realistic evaluations. Innovation is essential in drug development – especially when existing medications are no longer effective, for example due to antibiotic resistance or in the case of cancer, or when no treatment is available at all, as is the case for some rare diseases.
Improving evaluation methods for AI predictions
In a second study, Torsten Schwede’s team addresses another critical challenge: how to reliably assess AI predictions in an era of increasingly large and complex models. Modern AI systems generate millions of predictions for structures of proteins and macromolecular complexes as well as ligand interactions, based on growing datasets and increasingly large experimentally determined reference structures. This makes it challenging to evaluate how reliable these predictions are.
In “Nature Methods”, the researchers identify limitations in existing structure comparison methods and present robust approaches to better assess the reliability of AI-generated predictions. Such tools will be essential for using AI in biomedical research and drug discovery.
Publications:
Peter Škrinjar, Jérôme Eberhardt, Gabriel Studer, Gerardo Tauriello, Torsten Schwede, Janani Durairaj. Evaluating generalization in protein-ligand cofolding methods. Nature Structural & Molecular Biology, published online 8 May 2026
Gabriel Studer, Xavier Robin, Stefan Bienert, Janani Durairaj, Peter Škrinjar, Gerardo Tauriello, Andrew Waterhouse, Torsten Schwede. Comparing macromolecular complexes - a fully automated benchmarking suite. Nature Methods; published online 22 December 2025
Contact: Communications, Katrin Bühler