
{kind=link}
Obwohl alle Zellen eines Organismus die gleiche genetische Information enthalten, ist jede auf ihre Weise individuell. Nicht nur die verschiedenen Zelltypen unterscheiden sich in ihrer molekularen Ausstattung, auch wenn einzelne Zellen auf Reize reagieren, sich an ihre Umwelt anpassen, bei Alter oder bei Krankheit verändert sich ihr Innenleben, ihre molekulare Beschaffenheit. Jede Zelle enthält Hundertausende von Boten-RNAs (mRNAs) für die tausenden von Genen im Erbgut. Dieses mRNA-Expressionsprofil liefert ein Abbild der individuellen Eigenschaften einer Zelle, von zellulären Programmen und Anpassungen.
mRNA-Signaturen für einzelne Zellen
Die Einführung der Einzelzell-RNA-Sequenzierung (scRNA-seq) vor einigen Jahren war für die Wissenschaft daher ein grosser Durchbruch, der viele Bereiche von der Entwicklungs- bis zur Infektionsbiologie beflügelt hat. Mit dieser Hochdurchsatztechnologie können Forschende heutzutage mRNA-Expressionsprofile, für viele tausende einzelne Zellen gleichzeitig erstellen. Die gewonnenen Daten geben ein sehr detailliertes Bild vom Zellgeschehen beispielweise während der Organentwicklung oder bei Krankheiten.
Fehlerhafte Datenanalyse mit gängigen Tools
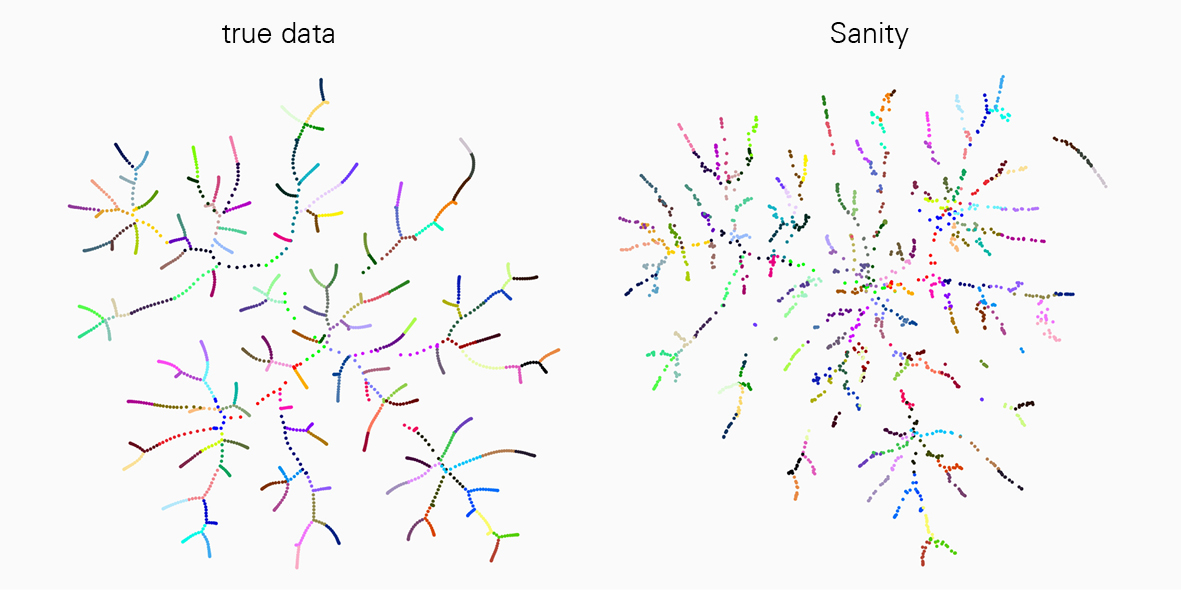
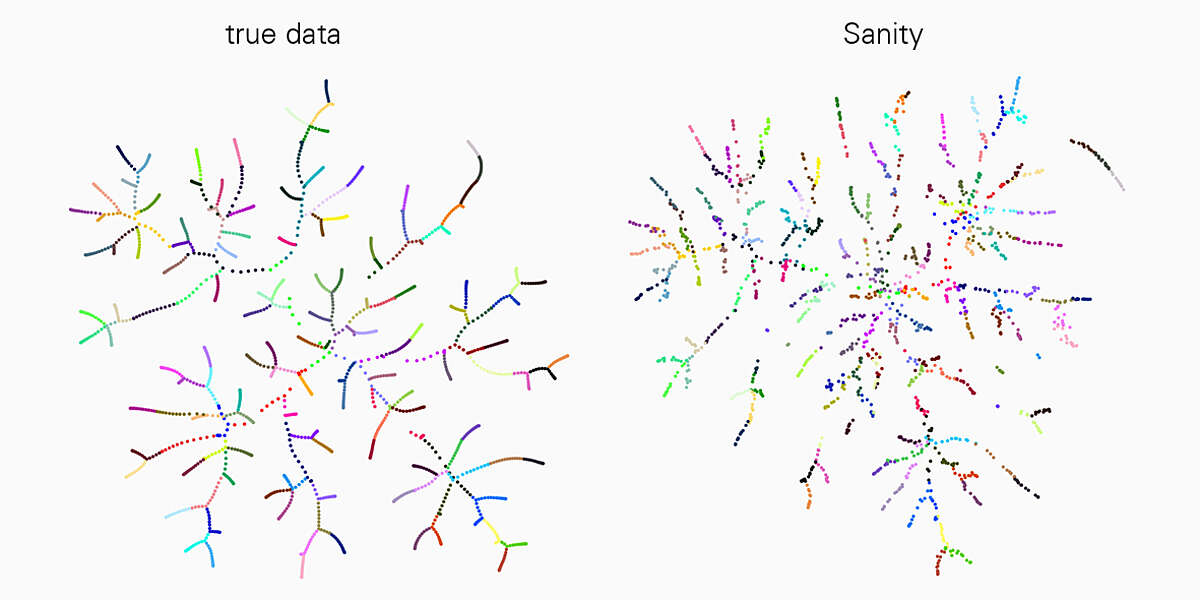
Eine grosse Herausforderung bei der Interpretation von scRNA-seq-Daten ist, dass die Messungen schwanken und dieses sogenannte «Rauschen» auf komplizierte Weise vom Zustand der Zelle abhängt. Die derzeit verfügbaren Tools ignorieren dieses Problem jedoch meist. «In unserer Arbeit konnten wir zeigen, dass viele der Tools zur scRNA-seq-Analyse die Daten erheblich verzerren und sogar verschiedene Artefakte produzieren», sagt Jeremie Breda, Erstautor der Studie. «Mit ‘Sanity’ haben wir nun erstmals eine effiziente Methode entwickelt, mit der sich das ‘Rauschen’ in den Messwerten, sei es durch die Messung selbst oder aufgrund biologischer Schwankungen, zuverlässig korrigieren lässt.»
Verlässliche Datenanalyse mit Sanity-Software
Diese Arbeit ist ein Meilenstein auf dem Gebiet und für alle Forschenden, die mit der scRNA-seq-Technologie arbeiten, ein grosser Fortschritt. «Wir haben ‘Sanity’ mit einigen vielfach genutzten Tools zur Datenanalyse verglichen und festgestellt, dass unsere Methode sehr verlässlich ist und die anderen in einigen Anwendungen wie der Identifizierung von unterschiedliche exprimierten Genen oder Subtypen von Zellen, übertrifft», erklärt van Nimwegen. Für das richtige Verständnis zellulärer Prozesse ist es entscheidend, dass die Daten zur Genexpression die biologische Wirklichkeit korrekt widerspiegeln.
Die Sanity-Software steht Forschenden weltweit unter github.com/jmbreda/Sanity zum Download zur Verfügung.
Originalpublikation:
Jérémie Breda, Mihaela Zavolan and Erik van Nimwegen. Bayesian inference of the gene expression states of single cells from scRNA-seq data. Nature Biotechnology; published online 29 April 2021
Kontakt: Kommunikation, Katrin Bühler