
Main Content
Proteomics at the Biozentrum
Proteomics rapidly evolves from a discovery-oriented technique to a robust and sensitive quantitative tool in biological research to study changes in protein expression and protein modifications in a high-throughput manner.
Proteomics service is available for all research groups of the Biozentrum
The Proteomics Core Facility (PCF) provides infrastructure for the identification and quantification of proteins and their modifications. This includes profound expertise in phosphopeptide enrichment strategies, various platforms for protein and peptide separations, state-of-the-art mass spectrometry (MS) for discovery based MS and LC-MS/MS experiments as well as directed and targeted MS workflows for sensitive and consistent quantitative monitoring of pre-selected sets of proteins. The PCF continuously develops and adapts new sample preparation techniques, MS approaches and software tools to provide optimal analytical services for the individual research groups and their projects.
Areas of interest
Besides the analytical service for the Biozentrum, research at the PCF focuses on the development and application of (i) quantitative phosphoproteomics for tracking complex cellular phosphorylation events (Jenoe lab) and (ii) data-independent MS workflows for proteome-wide quantitative studies of microbes and the specific monitoring of proteins and their modifications in complex systems such as human cell lines (Schmidt lab).
Specifically, snapshots of signaling pathways in hepatocellular carcinomas (HCC) are acquired by quantitatively comparing proteomes and phosphoproteomes of biopsies taken from HCC patients. The aim is to capture the proteome and the phosphorylation status of signaling pathways by analyzing serial biopsies of patients before and during treatment with the protein kinase inhibitor Sorafenib. Other projects focus on proteome-wide studies of various human pathogens, identification of protein-protein interactions and determining precise concentrations of outer membrane proteins using targeted proteomics assays. Furthermore, extensive quantitative datasets of human cancer cells showing chromosome instability (CIN) were acquired to identify key factors and cellular processes involved in the formation and tolerance of CIN.

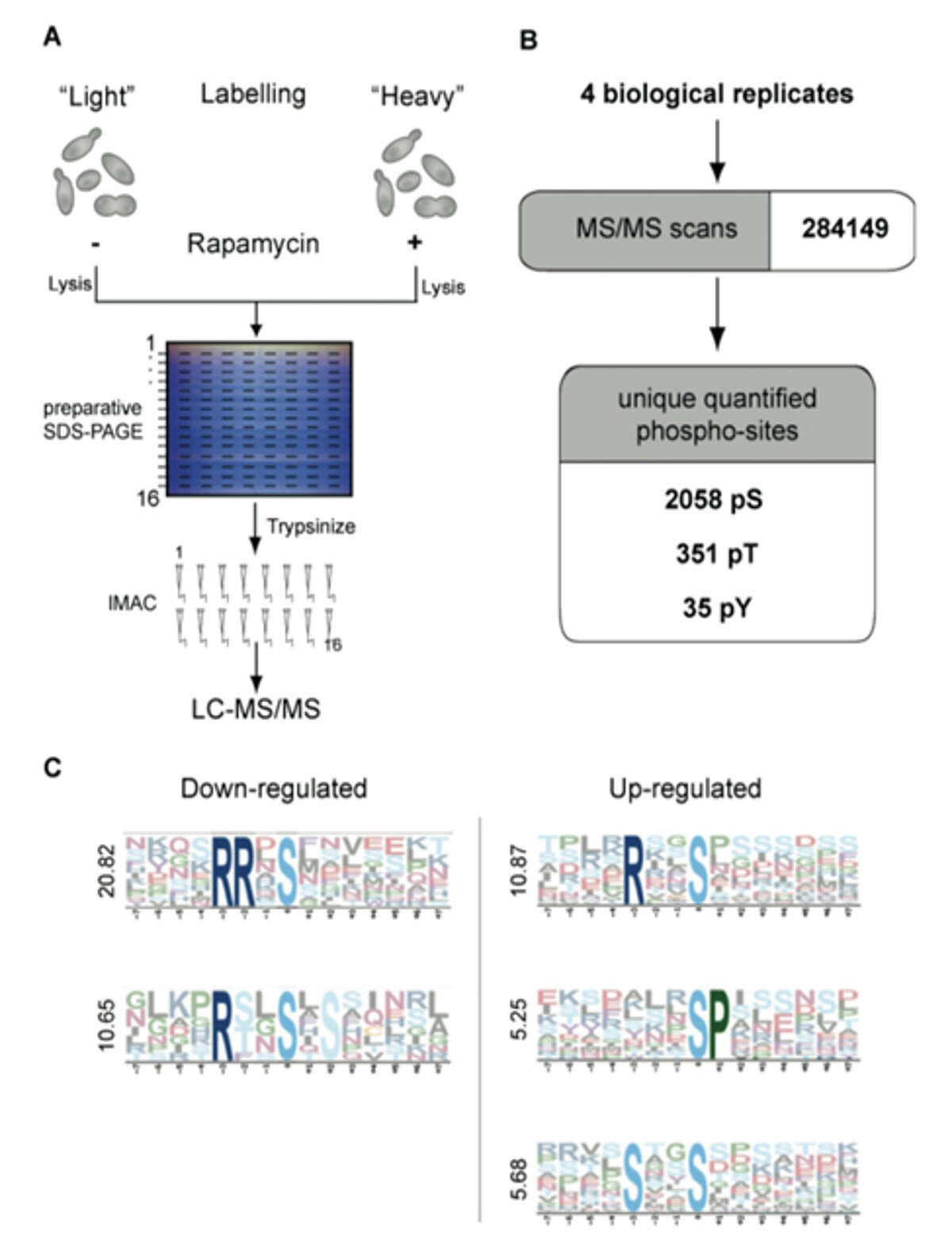
Fig. 1: Quantitative analysis of the rapamycin-sensitive phosphoproteome by SILAC. A) Two yeast cultures are metabolically labeled with normal or isotopically labeled Lysine and Arginine (heavy culture). The heavy culture is treated for 15 minutes with rapamycin. Cell lysates mixed in a ratio of 1:1 are separated by preparative SDS-PAGE, sliced into horizontal bands and proteins are digested. Phosphopeptides are enriched via IMAC and measured in an LTQ-Orbitrap. B) Four independent experiments yielded 972 phosphoproteins, corresponding to 2,383 unique phosphopeptides. C) Motif analysis with Motif-X of all down-regulated and up-regulated phosphopeptide sequences. Motifs are ranked from top to bottom according to their score.
{kind=link}
Areas of activity
The PCF supports projects requiring the identification and quantification of proteins and protein modifications. Preferably, the PCF should be involved at every stage: in the planning of new research projects, during the initial phase and while the project progresses. Only this allows the PCF to provide the best analytical tools at all stages of the project and to adapt the analytical strategies to the specific needs. Furthermore, this facilitates the interpretation of the data and its communication in a user-friendly and plain manner.
Specific services and resources
In detail, we provide the following state of the art MS instrumentation and methods for the research groups:
LC-MS/MS platforms:
- Two High-resolution hybrid LTQ Orbitrap-Elite coupled online to an Easy-nLC-system (both from Thermo-Fisher Scientific) for discovery-driven workflows
- TSQ Vantage Triple Stage Quadrupole Mass Spectrometer coupled online to an Easy-nLC-system (both from Thermo-Fisher Scientific) for hypothesis-driven workflows using selected-reaction monitoring for protein quantification
-
Q-Exactive HF coupled online to an Easy-nLC-system (both from Thermo-Fisher Scientific) for discovery-driven and data-independent MS workflows
Sample preparation and fractionation instruments:
- 3100 OFFGEL Fractionator for peptide separation using isoelectric focusing (Agilent)
- Capillary liquid chromatograph for peptide separation and fractionation (Agilent)
Software:
- Database search tools: Mascot, Sequest and XTandem for tandem mass spectra interpretation, also in combination with the trans proteomic pipeline
- Scaffold (Proteome software) for communicating proteomics results in a user-friendly format
- Progenesis QI label-free quantification software (Nonlinear Dynamics)
- MaxQuant for quantification of isotopically labeled samples
- SpectroDive/spectronaut/Skyline for analysis of data-independent protein quantification experiments
- In-house software tool SafeQuant for absolute protein quantification and statistical analysis of large quantitative datasets
Methods:
- Protein identification, including posttranslational modifications
- Absolute and differential protein quantification (label-free, isobaric isotope labeling-based)
- Enrichment and quantification of phosphopeptides
- Data-independent MS for consistent and accurate protein quantification in large-scale proteomics projects
Steering board
To coordinate Proteomics activities at the Biozentrum a PCF steering board was established in 2010. The steering board committee is composed of four group leaders who are strongly committed to proteomics, the Head of Administration of the Biozentrum and the two co-directors of the PCF.
Outlook
With the continuing advances in MS instrumentation (and the purchase of the new QE-HF), analytical methodology and software tools, proteomics is well suited to meet the requirements for modern biological projects on a system-wide level. In particular, data independent (DIA) proteomics workflows have great potential in reproducibly and extensively quantifying entire proteomes over large sample numbers. The new LC-MS platform is ideally suited for this type of analysis, which is currently integrated and tested for several in-house projects. We will go one step further and develop optimized protocols for targeted quantification of post-translational modifications (PTMs), like phosphorylation and acetylation, as well as protein isoforms directly from cell lysates without the need for enrichment steps and considerably reducing required sample amounts. Besides, we are currently establishing a new solid phase extraction protocol that only requires minimal sample load and is particular useful for precious samples that are in short supply.

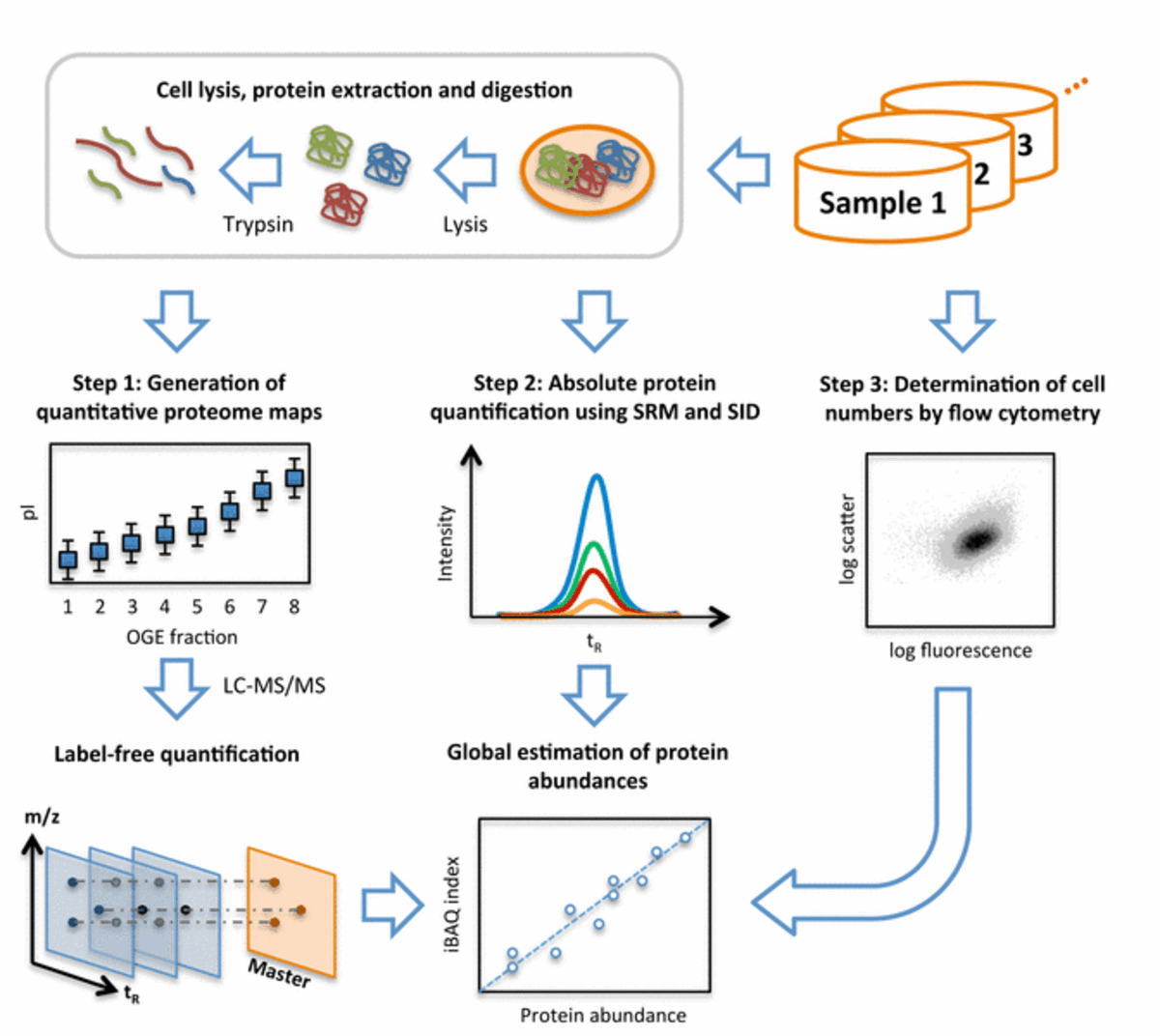
Fig. 2: Workflow of system-wide protein abundance determination. The workflow comprised three steps. First, cells of the various samples were lysed, proteins extracted and proteolyzed using trypsin. The peptide mixtures were either further fractionated using OFFGEL electrophoresis (OGE) or directly analyzed in biological triplicates by shotgun LC-MS/MS and quantified by label-free quantification. Second, the cellular concentrations of 41 proteins covering all components of the glycolysis pathway were determined across all samples by selected-reaction monitoring (SRM) and stable isotope dilution (SID). Therefore, for each protein, heavy labeled reference peptides (selected from the shotgun LC-MS/MS experiment) were synthetized. After spiking known amounts of these references into each sample, absolute quantities were determined for the corresponding proteins by SRM. Third, the numbers of cells taken for LC-MS/MS analyses were determined for each sample by flow cytometry. With this information, the protein concentrations determined by SRM/SID could be transformed to protein copies/cell and a quantitative model was built to translate MS-intensities of all quantified proteins to cellular abundance estimates using the Intensity Based Absolute Quantification (iBAQ) approach.
{kind=link}