
Main Content
Regulators of gene expression from stem cell differentiation to aging
Our group is interested in the mechanisms that control gene expression from very early to very late in human life. In eukaryotes, gene expression is a complex process, with many steps, all amenable to regulation. We are interested primarily in mechanisms of post-transcriptional gene expression control, involving small non-coding RNAs and RNA-binding proteins.
From binding motifs to functional impact

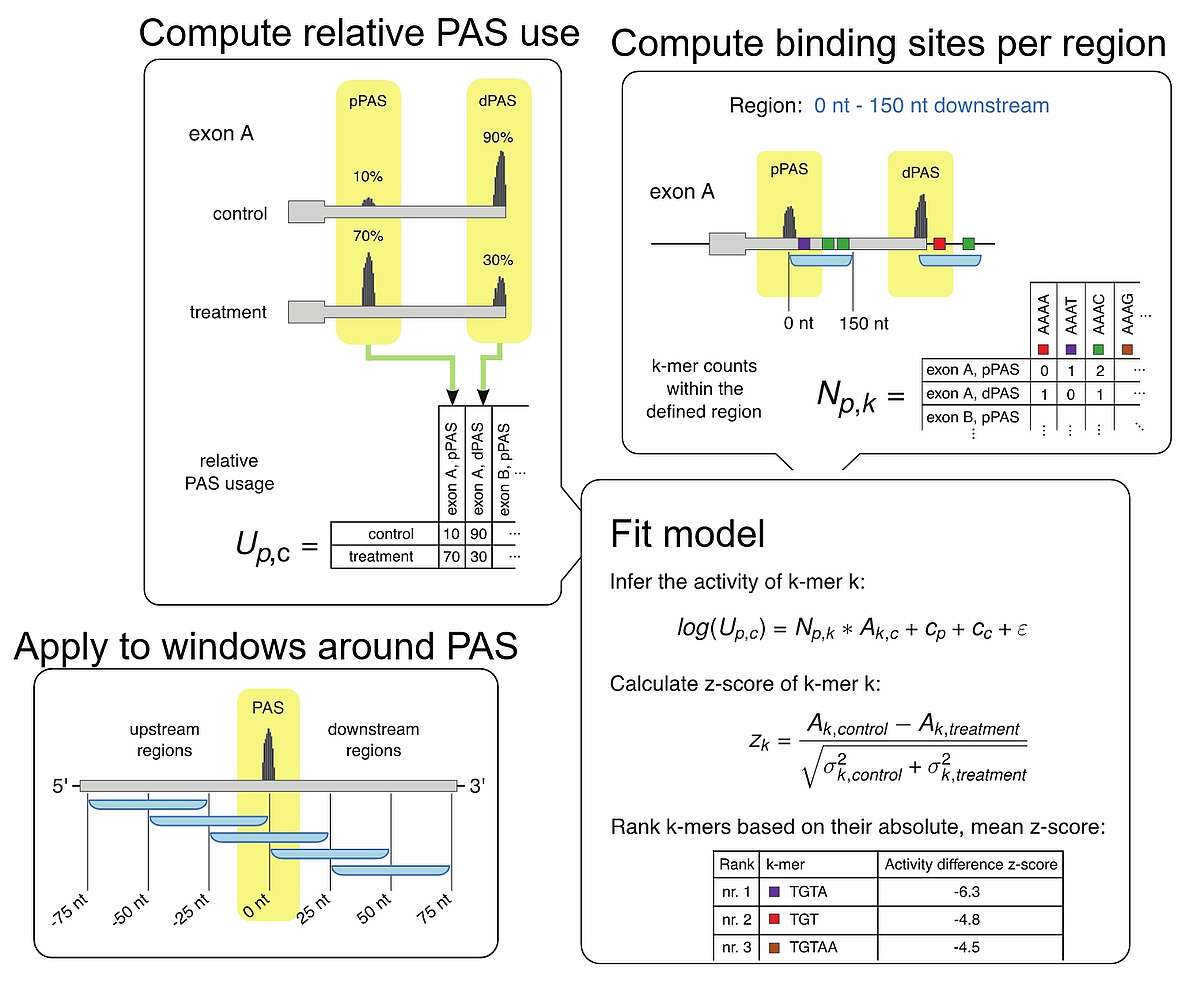
Fig.1. KAPAC model for inferring the impact of RNA-binding proteins on polyadenylation. Based on either 3’ end or full-length RNA sequencing data, we infer the relative usage of each poly(A) site in a given terminal exon. Then, we determine the number of binding sites for specific RBPs or the counts of all possible k-mer sequences within a window around each quantified poly(A) site. We then fit a linear model describing the log-usage of poly(A) sites in terms of the RBP binding sites or sequence motifs that they contain, obtaining estimates of activities of sequence motifs of polyadenylation. By carrying out the analysis to different windows around poly(A) sites we can detect position-specific impact of RBPs on polyadenylation.
The production, processing and degradation of RNAs is modulated by various protein complexes that recognize short sequence motifs in their targets. Thus, a prerequisite for understanding why the level of various RNAs varies between conditions is to know with which complexes these RNAs interact. Our group develops computational methods to infer binding sites for various regulators across the transcriptome and to link these interactions to specific functional outcomes, such as isoform selection and transcript degradation.
The two main classes of RNA regulators are miRNAs and RNA-binding proteins. To be able to predict binding sites for these regulators genome-wide, we need to know their sequence specificity. In the past decade, methods have been perfected for the high-throughput isolation of binding sites of these regulators. Building on such data, we have proposed the first biophysical model of miRNA-target RNA interaction, a model that predicts the affinity of such interactions with high accuracy. For RNA-binding proteins (RBPs), both in vivo and in vitro binding data has been generated, but principled models describing the sequence specificity of RBPs are yet to be developed. We are currently pursuing such efforts.
Knowing where RBPs bind gives us the opportunity to investigate the impact of these interactions on the target RNAs. To this end, we develop models to explain various properties of RNAs, such as the usage of specific processing sites or the expression level of individual isoforms in terms of the combination of binding sites that these RNAs have for various regulators. For example, we developed the K-mer activity on poly(A) site choice (KAPAC) model for inferring sequence motifs that impact the choice of polyadenylation sites. This enabled us to identify novel regulators of RNA processing. We are currently extending KAPAC to work with a more general representation of binding sites and we are developing an analogous method for the identification of splicing regulators.
Regulation of isoform expression via alternative polyadenylation

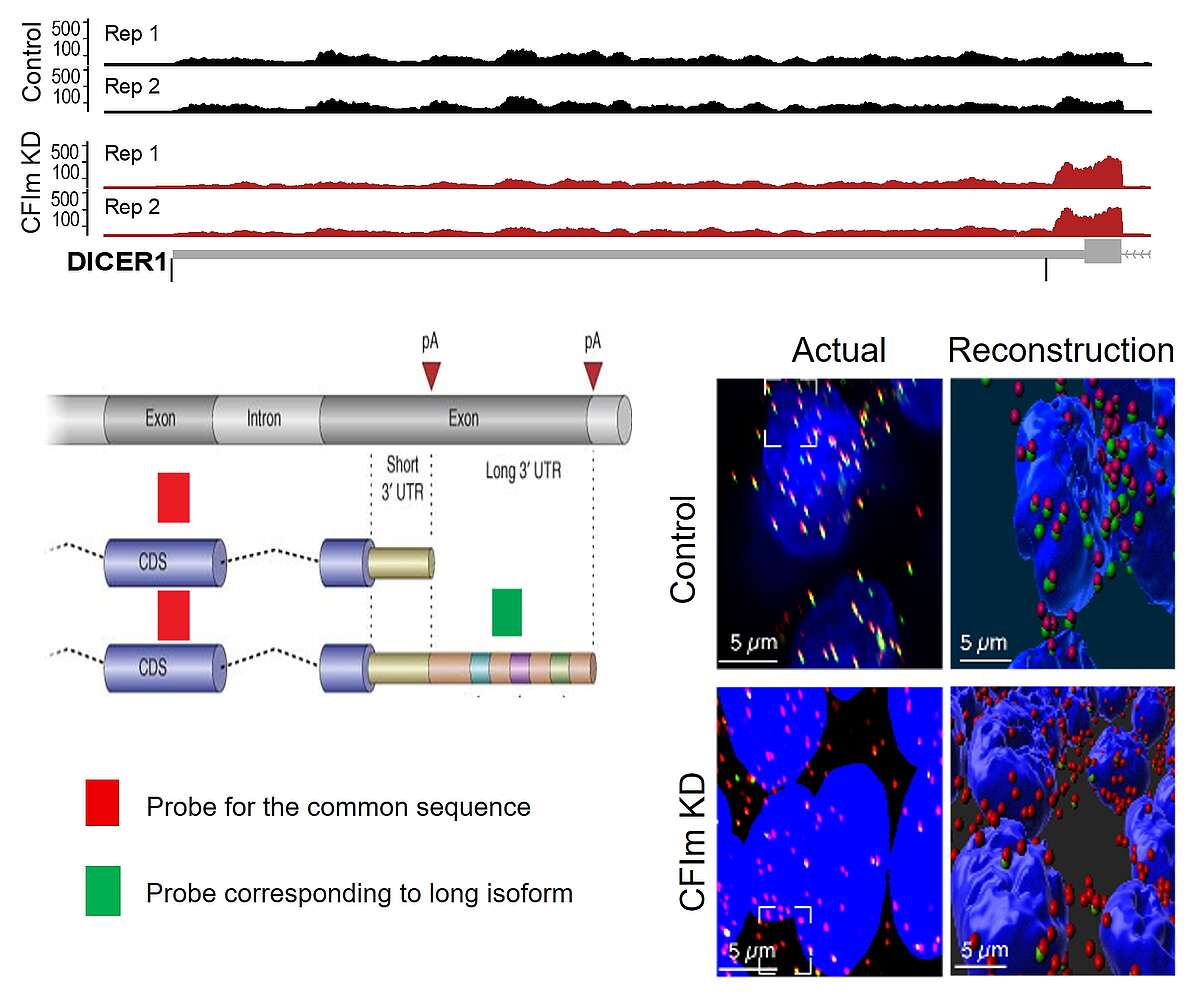
Fig. 2. 3’UTR shortening induced by the knockdown of the 3’ end processing factor CFIm. Shown is a schema of the last 2 exons of a gene, the terminal exon having two potential poly(A) sites. The 3’ end processing at the coding-region proximal site leads to a short 3’UTR isoform, while the processing at the distal site generates a long 3’UTR isoform. We have designed a probe for the coding region of the Dicer1 gene (in red), probe which binds to both the short and the long 3’ UTR isoform, as well as a probe for the region that is unique to the long 3’UTR isoform (in green). The figure shows HEK293 cells, nuclei in blue, with either normal expression of CFIm (top) or reduced expression (bottom). There is a strong depletion of the green probe signal in the bottom panels, indicating that the long 3’UTR isoform expression is much reduced in cells with low CFIm expression.
Although the number of genes in worm and human cells is not very different, the number of variant transcripts that are generated from a given gene in human cells is much larger than in worms. The main source of variation comes from the multiplicity of sites where the transcription of a gene can be initiated as well as of sites where the transcripts undergo 3’ end cleavage. We have discovered a number of proteins that contribute to the choice of cleavage site, most prominently a subcomplex of the core 3’ end processing machinery, the mammalian cleavage factor I (CFIm). CFIm is the main determinant so far described of 3’UTR length, reduced expression of CFIm leading to drastic shortening of 3’UTRs. In current experimental work we are characterizing the main CFIm targets and the way they impact cellular processes such as signaling, stress responses and miRNA-dependent regulation.
Interestingly, 3’UTRs are systematically shorter in tumors compared to normal tissue, for reasons that are not yet known. To study this process, we have developed a method to analyze the dynamic of 3’UTR length in individual cell types within and outside of tumor microenvironment, based on single cell RNA sequencing (https://github.com/zavolanlab/SCUREL). We found that 3’UTR shortening occurs in all cell types within the tumor microenvironment, and is not restricted to cancer cells. The targets of this process, while not identical among cell types, encode proteins that are involved in various aspects of protein metabolism.
We also work on developing resources to study alternative polyadenylation. Most prominently, we developed and maintain the PolyASite database (polyasite.unibas.ch) of polyadenylation sites obtained by methods specifically designed for this purpose (3’ end sequencing). The resource facilitates efficient finding, exploration and export of poly(A) sites, and includes estimates of the relative use of each site across all of the analyzed samples. Furthemore, we contribute to APAeval, a community effort to evaluate computational methods for the detection and quantification of poly(A) sites and the estimation of their differential usage across RNA-seq samples.
Mechanisms of translational control
With the emergence of the ribosome footprinting method, it became clear that gene expression is also extensively regulated at the step of protein synthesis by ribosomes. Prompted by our finding that the expression of ribosomal protein paralogs varies substantially between cell types, to the extent that it allows one to classify hematopoietic cells, we have started to investigate how ribosomes can impart transcript-dependent rates of protein synthesis. Furthermore, we work on reconstructing the translation landscape of various cell types and tissue, exploring the dynamic of this landscape as stem cells differentiate, organisms age, and normal cells undergo malignant transformation.
The aging mouse
A hallmark of aging is the perturbation of protein synthesis. Indeed, in an initial study that we carried out in yeast, we found that the main determinant of yeast replicative life span is the protein synthesis rate, regardless of the signal that modulates this rate. This led us to establish a collaboration with the Ruegg group at the Biozentrum to investigate the mechanism underlying the perturbed protein synthesis during mouse aging. A key aspect of the study was that we included models in which aging was known to be mitigated, e.g. by treating the mice with rapamycin or caloric restriction (2021, 2020). The study resulted in a wealth of data so far pertaining to the skeletal muscle (sarcoatlas). We are now extending this study to include not only other tissues, but also a combined treatment, with rapamycin and caloric restriction, as we found that these individual treatments reveal somewhat distinct sets of targets. We carry out a multi-omic analysis of samples, involving RNA-seq, proteomics, phospho-proteomics, and ribosome profiling, as well as modeling of translation and metabolism, to identify the key factors driving the progressive loss of function of organs with age. As one of the hypotheses about the emergence of aging in evolution is that it is a side effect of mechanisms evolved to prevent malignant transformation, we carry out similar molecular analyses in cancer cells.

Fig. 3. Schematic view of the “aging mouse” project. Mice were maintained from adult to advanced age under standard laboratory conditions, or under treatment with either rapamycin or caloric restriction. Samples were then collected to analyze via RNA sequencing, mass spectrometry and ribosome footprint sequencing. The computational analysis revealed molecular pathways that are affected in either of the three conditions relative to adult mice, and in the majority of cases, the changes brought by aging were reverted by the treatments. However, rapamycin had additional effects on metabolism and caloric restriction on protein synthesis. The model suggested by these results is shown in the right panel.
Automation, reproducibility, open science
The volume of data generated in a study has increased immensely in the past decade. Thus, we are undertaking efforts to facilitate the transparent, reproducible and efficient analysis of high-throughput data sets. Probably the most broadly used high-throughput technique is RNA sequencing. Although RNA-seq is employed in the large majority of molecular biology studies, the molecular biologists do not yet have sufficient tools to assess the quality of their data and obtain a basic summary that can inform subsequent analyses and experiments. To meet this need we have developed ZARP (Zavolan-Lab Automated RNA-Seq Pipeline), a generic RNA-Seq analysis pipeline that allows users to process and analyze Illumina short-read sequencing libraries with minimum effort. ZARP combines publicly available bioinformatics tools according to our extensive experience in the analysis of RNA-seq data, generating reports of sample quality metrics as well as functional annotation.
We are also working on setting up a workspace (frontend and backend) that allows users to run computational analysis workflows (Snakemake and Nextflow in particular) on the sciCORE infrastructure, based on community-wide standards set by the Global Alliance for Genomics and Health.


{kind=link}
{kind=link}