
{kind=link}
Although the genetic information is identical in all cells of an organism, each cell takes on its own individual state. Not only do cells come in various cell types that differ in their molecular makeup, but individual cells respond to stimuli, adapt to their specific environment, and as they change with age or disease, their inside, i.e. their molecular composition, also changes. Each cell contains hundreds of thousands of messenger RNA (mRNA) for the thousands of genes in the genome, and this mRNA expression profile provides an image of the individual characteristics of a cell, reflecting both cellular programs and adaptations.
mRNA signatures for single cells
The introduction of single-cell RNA sequencing (scRNA-seq) some years ago, was a major breakthrough for science, spurring a wide range of research fields from developmental to infection biology. Today, this high-throughput technology enables researchers to generate mRNA expression profiles, for many thousands of individual cells simultaneously. The acquired data allows for a highly detailed picture of cellular processes, for instance during organ development or disease processes.
Incorrect data analysis with commonly used tools
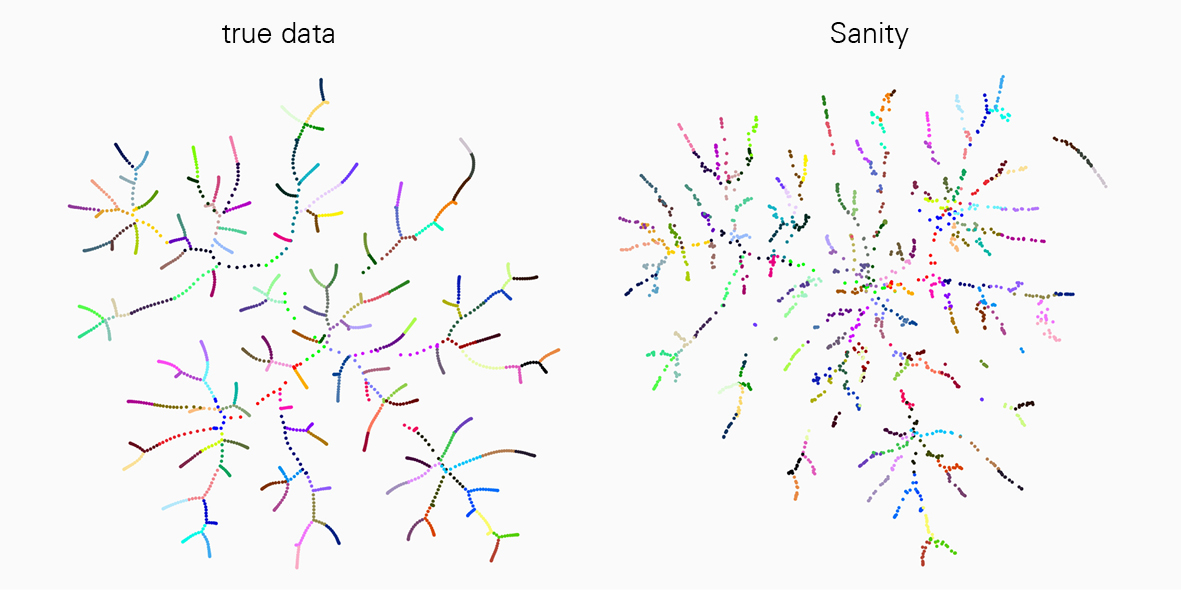
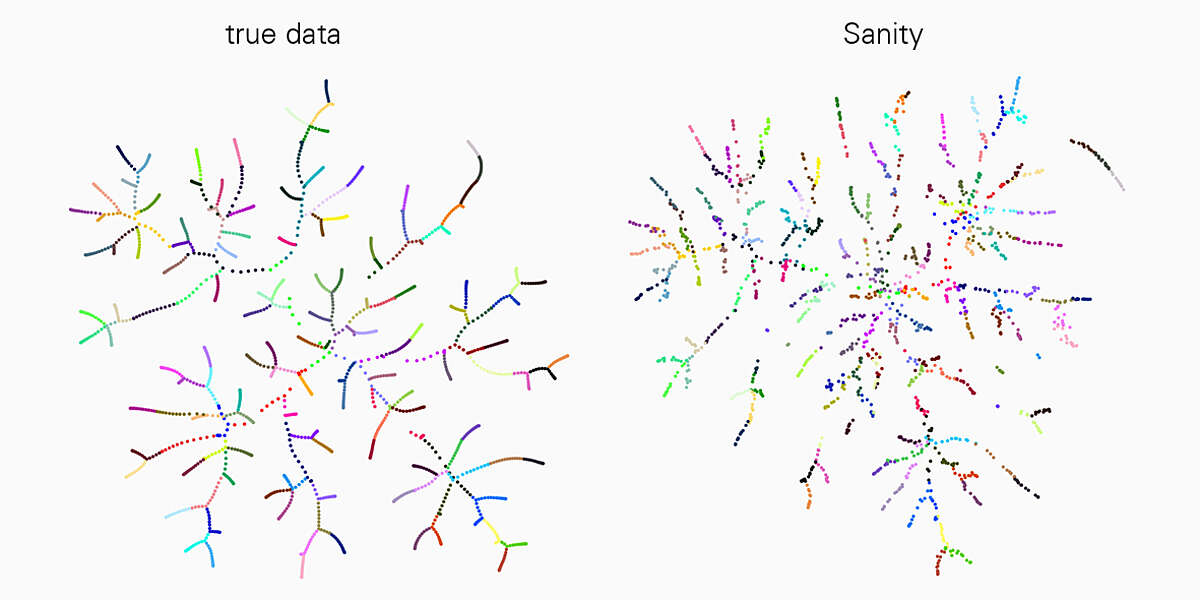
A major challenge to interpreting scRNA-seq data is that the measurements are very noisy, and the noise properties depend on the state of the cell itself in a complex manner. However, existing analysis tools mostly ignore these complications. “In our current work we have been able to demonstrate that many of the popular tools for scRNA-seq analysis severely distort the data and even produce various artefacts,” says Jeremie Breda, first author of the study. “With ‘Sanity’ we have now developed a rigorous method for reliably correcting both biological and measurement noise.”
Reliable data analysis using Sanity software
This work is a landmark in the field and a great advancement for all researchers working with scRNA-seq technology. “We compared ‘Sanity’ with several widely used tools for data analysis and showed that our method is very reliable and outperforms others in various applications such as in identifying differently expressed genes or clustering into cell subtypes,” explains van Nimwegen. "To properly understand cellular processes, it is crucial that our analysis of gene expression data correctly reflects the underlying biological reality."
The Sanity software is freely available to researchers worldwide and can be downloaded at: github.com/jmbreda/Sanity
Original publication:
Jérémie Breda, Mihaela Zavolan and Erik van Nimwegen. Bayesian inference of the gene expression states of single cells from scRNA-seq data. Nature Biotechnology; published online 29 April 2021
Contact: Communications, Katrin Bühler