
Main Content
Nuclear magnetic resonance spectroscopy of biomolecules
We apply and develop high-resolution Nuclear Magnetic Resonance (NMR) methods to elucidate structure, function, and dynamics of biological macromolecules. The structural and functional projects currently encompass the β1-adrenergic G-protein-coupled receptor (GPCR); the interactions of the GPCR CCR5, which is also the HIV1-coreceptor, with its chemokine ligand RANTES; Abelson kinase, a prime drug target in the treatment of chronic myelogeneous leukemia;, bacterial PilZ domains, which are targets for signaling via cyclic di-GMP; lipopolysaccharide, the causative agent of endotoxic shock; the TipA multidrug resistance protein of Streptomyces lividans; and an atomic-detail description of the unfolded states of proteins and the protein folding transitions by new NMR methods.

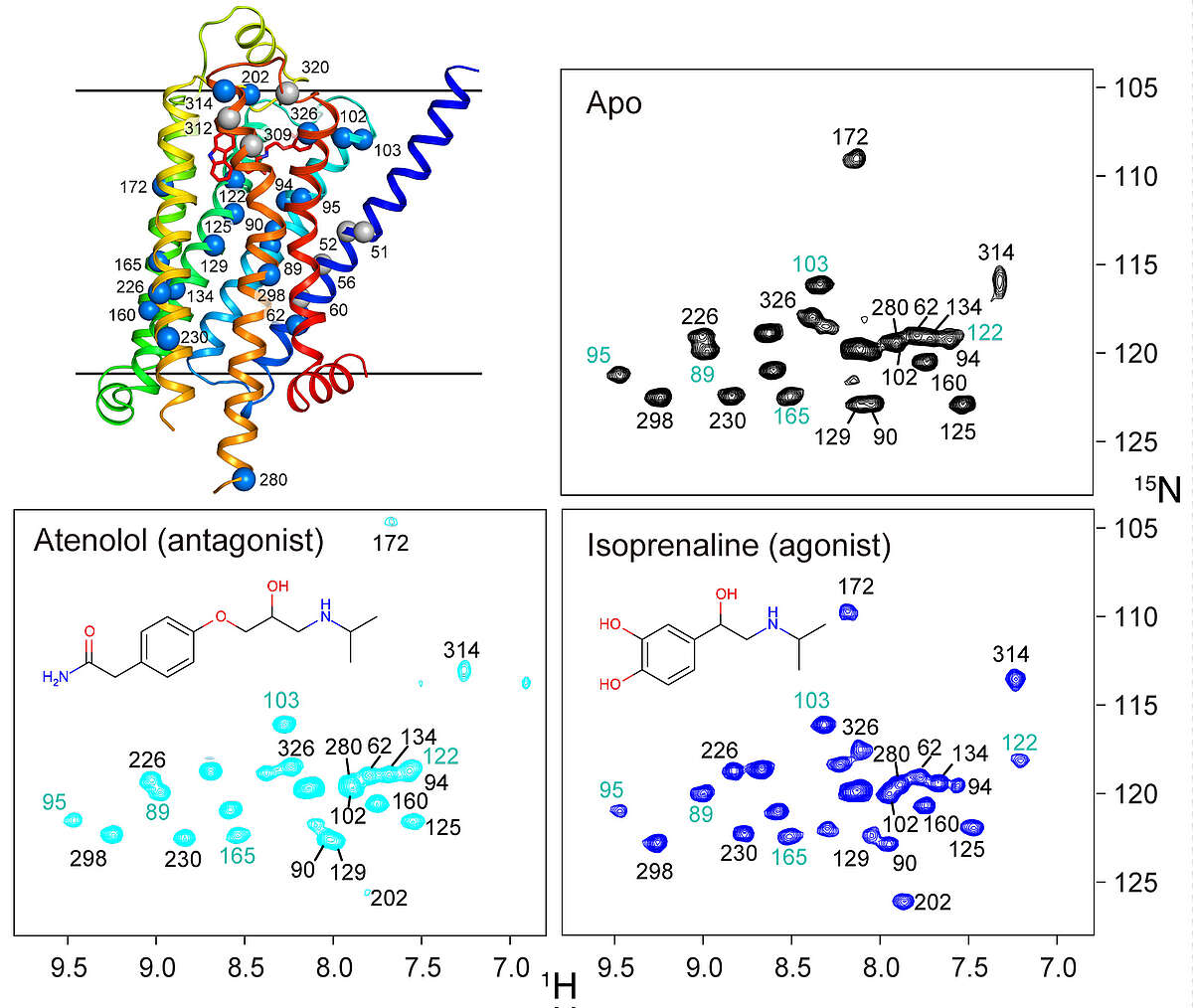
β1-adrenergic receptor. G protein-coupled receptors (GPCRs) are physiologically important transmembrane signaling proteins that trigger intracellular responses upon binding of extracellular ligands. Despite breakthroughs in GPCR crystallography, the details of ligand-induced signal transduction are not well understood due to missing dynamical information. We could recently show that receptor motions can be followed at virtually any backbone site in a thermostabilized mutant of the turkey β1-adrenergic receptor using NMR spectroscopy in solution [Isogai et al. Nature, 530, 237-241 (2016)]. Labeling with 15N-valine in a eukaryotic expression system provides over twenty resolved resonances that report on structure and dynamics in six ligand complexes and the apo form. The response to the various ligands is heterogeneous in the vicinity of the binding pocket, but gets transformed into a homogeneous readout at the intracellular side of helix 5 (TM5), which correlates linearly with ligand efficacy for the G protein pathway. The effect of several pertinent, thermostabilizing point mutations could be followed by reverting them to the native sequence. The results show hat even the fully thermostabilized receptor undergoes activating motions in TM5, but the fully active state is only reached in presence of Y227 and Y343 by stabilization with a G protein-like partner. The combined analysis of chemical shift changes from the point mutations and ligand responses identifies crucial connections in the allosteric activation pathway and presents a general experimental method to delineate signal transmission networks at high resolution in GPCRs. We are currently applying this technique to other GPCRs such as CCR5.

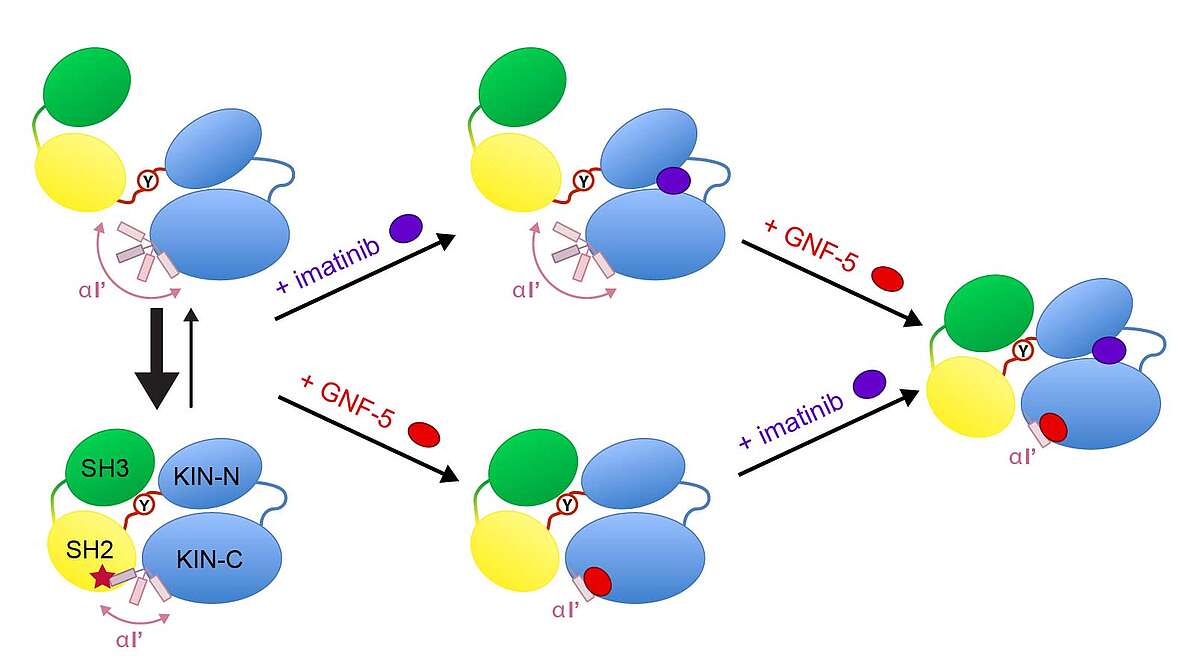
Abelson (Abl) kinase is a drug target in the treatment of chronic myelogenous leukemia (CML), against which clinically highly efficacious ATP-competitive inhibitors (imatinib and others) have been developed. However, spontaneous mutations in advanced-stage patients render these inhibitors inefficient.
Recently, a new type of allosteric inhibitors was shown in preclinical studies to overcome the resistance against ATP-binding pocket inhibitors [Zhang et al., Nature, 463, 501–506, (2010)]. The exact mechanism of the allosteric inhibition is currently unclear. In collaboration with Novartis (Basel) we had previously determined the unknown solution conformations of the Abl kinase domain alone [Vajpai et al., JBC, 283, 18292-18302 (2008)]. We have now also determined the solution conformations of a much larger 52-kDa SH3/SH2/kinase domain construct under the influence of various inhibitors by solution NMR and SAXS [Skora et al., Proc Natl Acad Sci USA 110, E4437–45 (2013)]. The addition of imatinib induces a large structural rearrangement characterized by the detachment of the SH3-SH2 domains from the kinase domain and the formation of an “open” inactive state, which is inhibited in the ATP site. In contrast to imatinib, binding of the allosteric inhibitor GNF-5 keeps the protein in the “closed” state. Combination of imatinib with GNF-5 brings the conformation again to a “closed” state. These findings on the allosteric actions of the two classes of inhibitors reveal molecular details of their recently reported synergy to overcome drug resistance.

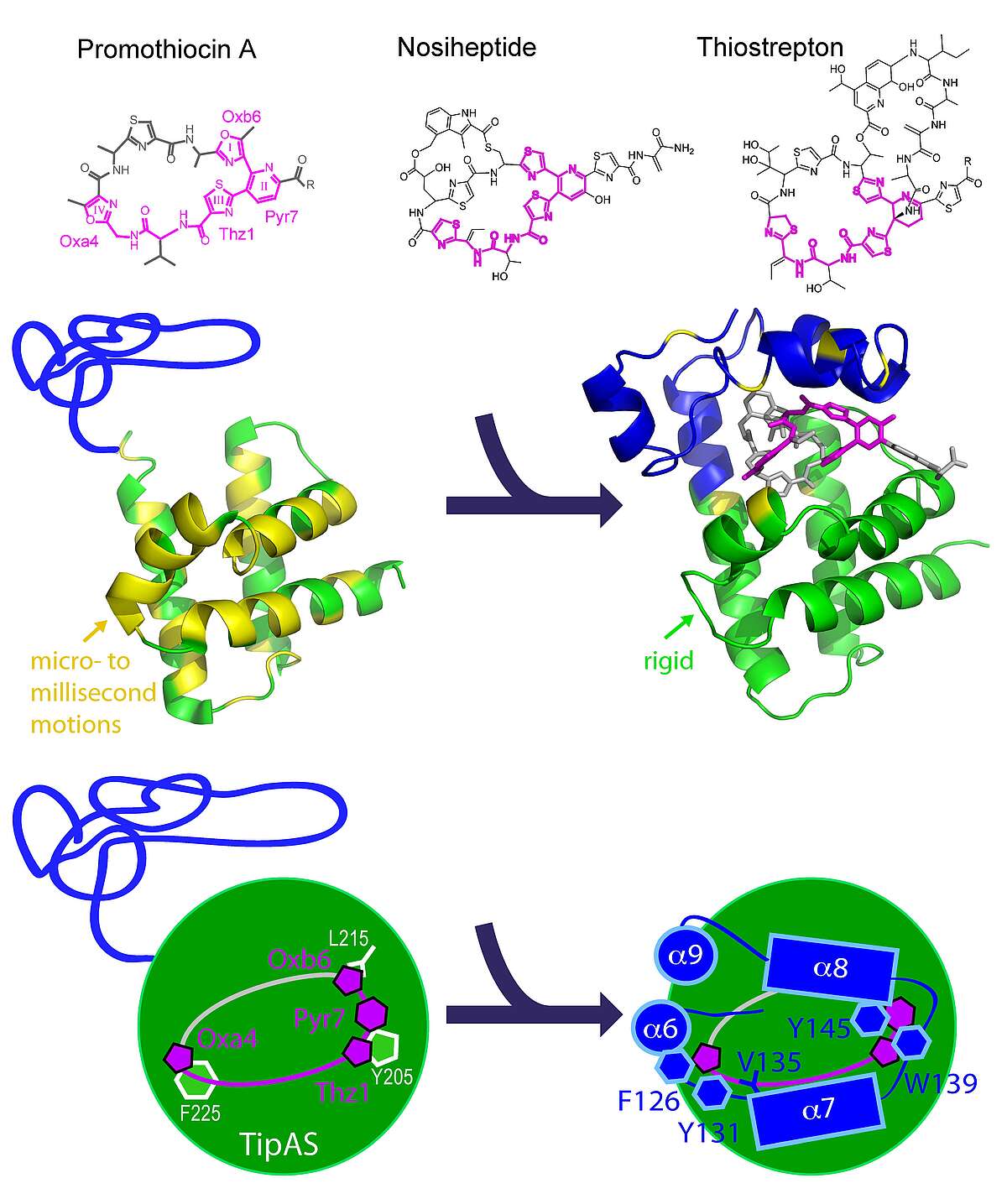
Multidrug Recognition. Thiostrepton-induced protein A (TipA) is a minimal bacterial, multidrug resistance (MDR) protein against very diverse antibiotics of the thiostrepton class. We have determined the solution structures and dynamics of the antibiotic binding domain of TipA in complexes with several antibiotics [Habazettl et al., PNAS, Dec. 8, 2014]. The data give unique insights into multidrug recognition by a complete MDR system: (i) a four-ring motif present in all known TipA-inducing antibiotics is recognized specifically by conserved TipA amino acids and induces a large transition from a partially unfolded to a globin-like structure. (ii) The variable part of the antibiotic is accommodated within a flexible cleft that rigidifies upon drug binding. Remarkably, the identified recognition motif is also the major interacting part of the antibiotic with the ribosome. Hence the TipA multidrug resistance mechanism is directed against the same chemical motif that inhibits protein synthesis. This makes the multidrug recognition mechanism by TipA a useful model for ribosomal thiopeptide binding and antibiotic development.

CCR5. The chemokine receptor CCR5 belongs to the class of G-protein coupled receptors (GPCRs). CCR5 is expressed on the surface of T-cells and activated after binding the endogenous chemokines MIP-1α, MIP-1β, and RANTES. CCR5 is the key molecule for HIV entrance into target cells, which proceeds via the sequential interaction of the viral protein gp120 with the host-cell factors CD4 and CCR5. Very promising HIV entry inhibitors are based on CCR5 ligands, comprising the natural ligand RANTES.
In recent years, we have obtained structural information on CCR5 and its complex with RANTES by studying the interaction of the soluble protein RANTES with peptides derived from the extracellular surface of CCR5 [Duma et al. J Mol Biol 365, 1063–1075 (2007)]. This information helped to devise more potent peptide-based HIV entry inhibitors [Chemistry & Biology 19, 1579-1588 (2012)]. As part of these efforts, we have recently characterized the dynamics, oligomeric states and detergent interactions of 5P12-RANTES, an engineered RANTES variant that is currently in phase I clinical trials [Wiktor et al,Biophys J, 105, 2586-97 (2013)]. We have also developed methods to produce CCR5 in sufficient amounts for structural and biophysical studies [Nisius et al. Protein Expr Purif 61, 155-162 (2008); Van den Bergh et al. PLoS One. 2012;7:e35074; Wiktor et al. J. Biomol. NMR 55, 79–95 (2013)].
This project emerged from the EU-FP7 project CHAARM (Combined Highly Active Anti-Retroviral Microbicides), a collaborative effort to develop combinations of new and existing anti-HIV agents, which can be applied topically to reduce transmission of HIV.

c-di-GMP signaling. Cyclic di-guanosine-monophosphate (c-di-GMP) is a bacterial signaling molecule that triggers a switch from motile to sessile bacterial lifestyles. This mechanism is of considerable pharmaceutical interest, since it is related to bacterial virulence, biofilm formation and persistence of infection. Understanding this mechanism may offer new routes to treatment of bacterial infections. We have recently solved the structure of the PilZ homolog PA4608 in complex with cyclic di-GMP [Habazettl et al. J Biol Chem 286, 14304 (2011)]. This complex shows large structural changes relative to the apo form. As a result of the rearrangements of N- and C-termini, a highly negative surface is created on one side of the protein complex. We propose that this movement of the termini and the resulting negative surface forms the basis for downstream signaling. We have recently also determined the exchange kinetics and equilibrium constants of various oligomeric forms of c-di-GMP [Gentner et al. J Am Chem Soc 134, 1019 (2012)]. These projects are in collaboration with the groups of Prof. Urs Jenal and Prof. Tilman Schirmer (Biozentrum).
Towards an atom-scale description of order in unfolded proteins from new NMR parameters. A detailed, quantitative description of the unfolded state ensemble of proteins is crucial for understanding protein folding, protein misfolding diseases such as Alzheimer’s and Parkinson’s, and function of intrinsically disordered proteins. The astronomical size of the conformational space of an unfolded polypeptide chain makes such a description both experimentally and theoretically very difficult.
Using new NMR experimental parameters comprising residual dipolar couplings and paramagnetic relaxation enhancements, we have been able to obtain a highly detailed, quantitative description of unfolded polypeptides. The results show that unfolded states contain considerably more residual, native-like structure than previously anticipated, thereby resolving Levinthal’s paradox that protein folding would need almost infinite times in an unbiased search of all accessible conformations.

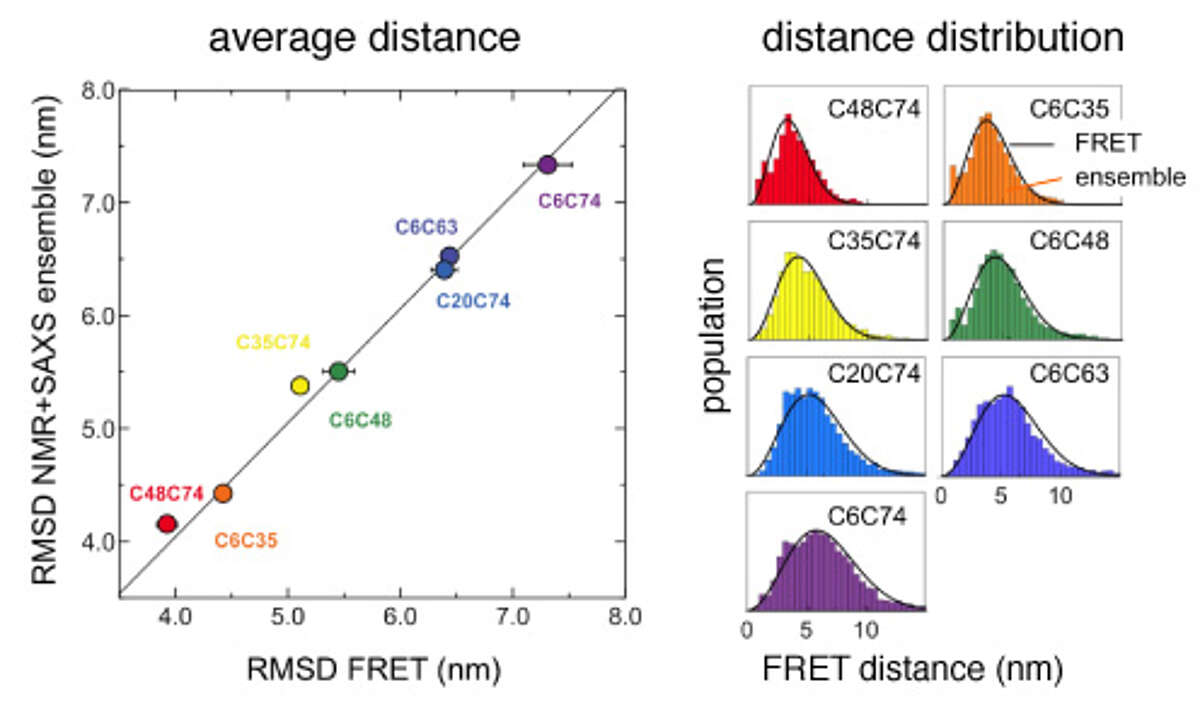
Comparison of distances between amino acids in urea-denatured ubiquitin that were derived either from a calculated structural ensemble based on NMR and SAXS data or from single molecule FRET experiments. Both the average distances (left) as well as the underlying distance distributions (right) agree exceedingly well.
Comprehensive structural and dynamical view of an unfolded protein from the combination of single-molecule FRET, NMR, and SAXS. We have determined structural ensembles of urea-denatured ubiquitin based on a very high number (>10/residue) of experimental (RDC, PRE, SAXS) constraints, which show the existence of about 10% residual native structure under these conditions [Huang and Grzesiek J Am Chem Soc, 132, 694–705 (2010)]. We have recently extended this work by single molecule FRET studies in collaboration with Prof. B. Schuler (U. Zürich) [Aznauryan et al. Proc Natl Acad Sci USA 113, E5389–98 (2016)]. We find excellent agreement between NMR- and FRET-derived distances. The FRET data indicate a long-range Gaussian chain behavior and long-range reconfiguration times of 50-100 ns, which agree with a polymer Rouse model with internal friction. The local native, secondary structure propensities evident from the NMR data are completely compatible with this Gaussian long-range behavior. The long-range and long-time scale information from single molecule FRET is highly complementary to the short-range and fast-time scale NMR information.

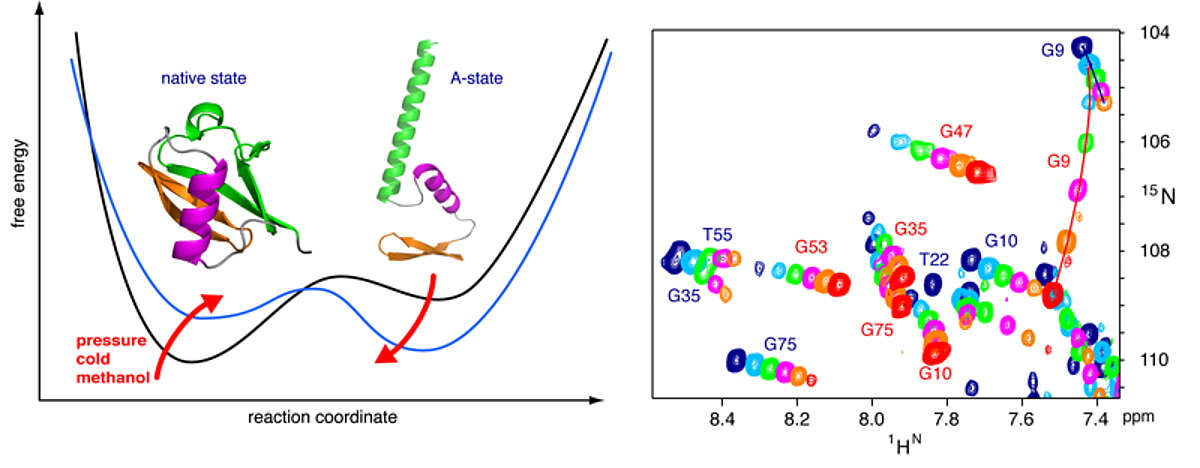
High pressure NMR reveals close similarity between cold and alcohol protein denaturation. Proteins denature not only at high, but also at low temperature as well as high pressure. These denatured states are not easily accessible for experiment, since usually heat denaturation causes aggregation, whereas cold or pressure denaturation occur at temperatures well below the freezing point of water or pressures above 5 kbar, respectively. We have obtained atomic details of the pressure-assisted, cold-denatured state of ubiquitin at 2500 bar and 258 K by high-resolution NMR techniques [Vajpai et al. Proc Natl Acad Sci USA 110, E368–76 (2013)]. This state contains on the order of 20 % native-like and non-native secondary structure elements. These structural propensities are very similar to the previously described alcohol-denatured (A-) state. The close similarity of pressure-assisted, cold-denatured and alcohol-denatured state supports a hierarchical mechanism of folding and the notion that similar to alcohol, pressure and cold reduce the hydrophobic effect. Indeed, at non-denaturing concentrations of methanol, a complete transition from the native to the A-state can be achieved at ambient temperature by varying the pressure from 1 to 2500 bar. This method should allow highly detailed studies of protein folding transitions in a continuous and reversible manner.

The application of high pressure at low temperature makes it possible to observe the cold-denatured state of ubiquitin by NMR at atomic resolution. This state contains native and non-native secondary structure elements that are every similar to the alcohol-denatured state. The transition can be studied in a continuous manner from the folded to the unfolded state.
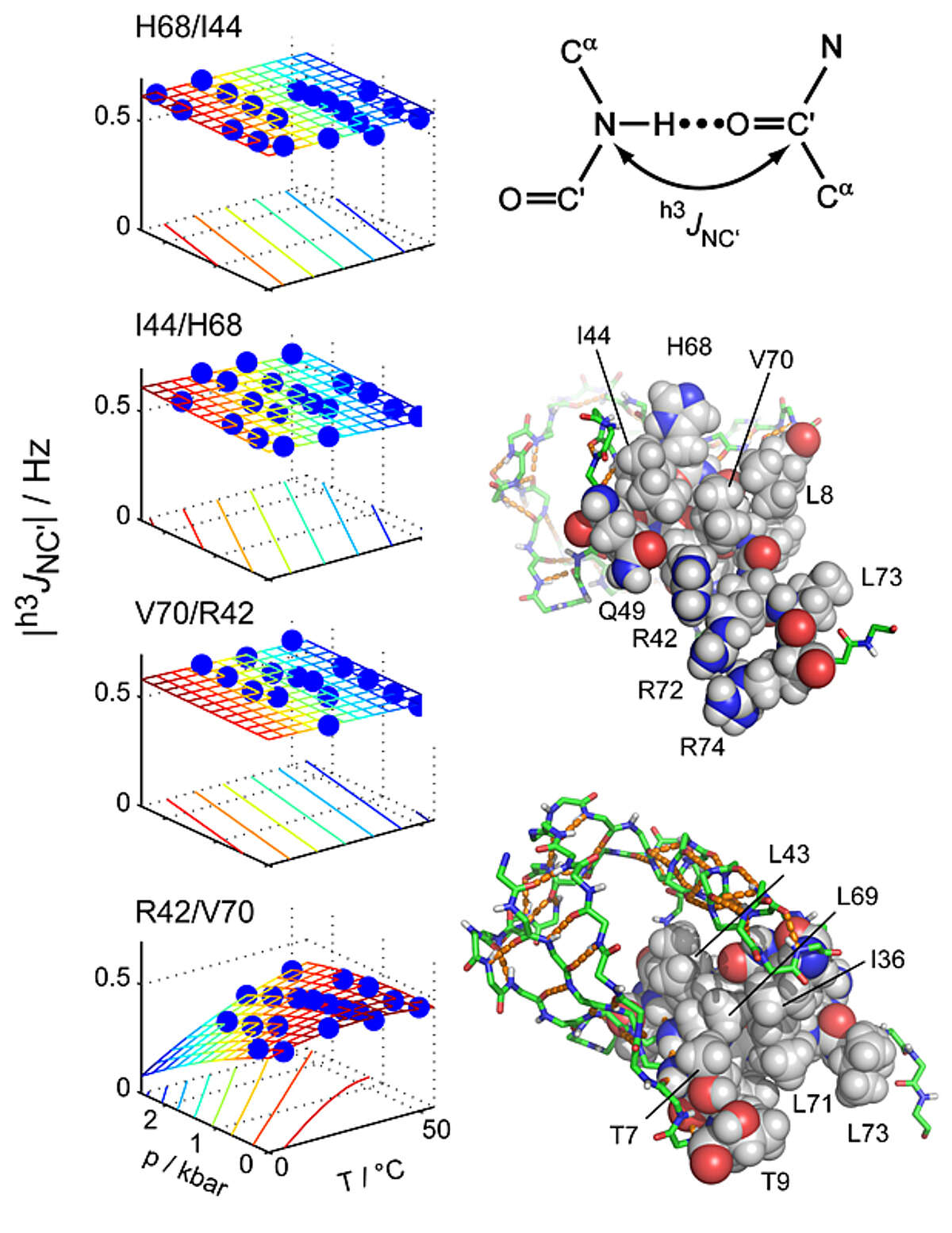
Key stabilizing elements of protein structure identified through pressure and temperature perturbation of its hydrogen bond network. Hydrogen bonds are key constituents of biomolecular structures, and their response to external perturbations may reveal important insights about the most stable components of a structure. NMR spectroscopy can probe hydrogen bond deformations at very high resolution through hydrogen bond scalar couplings (HBCs). However, the small size of HBCs has so far prevented a comprehensive quantitative characterization of protein hydrogen bonds as a function of the basic thermodynamic parameters of pressure and temperature. Using a newly developed pressure cell, we have mapped pressure- and temperature-dependent changes of 31 hydrogen bonds in ubiquitin by measuring HBCs with very high precision [Nisius and Grzesiek Nat Chem 4, 711–717 (2012)]. Short-range hydrogen bonds are only moderately perturbed, but many hydrogen bonds with large sequence separations (high contact order) show greater changes. In contrast, other high-contact-order hydrogen bonds remain virtually unaffected. The specific stabilization of such topologically important connections may present a general principle to achieve protein stability and to preserve structural integrity during protein function.
Important Partners
M. Blackledge, (Institut de Biologie Structurale, Grenoble, FR); S. Dames (Technical University, Munich, DE); L. Emsley (Ecole Normale Superieure de Lyon, FR); M. Geyer (Max-Planck-Institut für molekulare Physiologie, Dortmund, DE); O. Hartley (University of Geneva, CH); W. Jahnke (Novartis Pharma, Basel, CH); U. Jenal (Biozentrum); J. Lewandowski (University of Warwick, UK); G. Schertler (Paul-Scherrer Institut, Villigen); T. Schirmer (Biozentrum, Basel, CH); B. Schuler (University of Zürich, CH); H. Stahlberg (Biozentrum); C. Thompson (UBC, Vancouver, Canada); L. Vangelista (DIBIT, Milano, IT); U. Zähringer (Forschungszentrum Borstel, DE).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}