
Muscular dystrophies
Muscular dystrophies are rare, genetic diseases often caused by mutations in genes important for the structural stability of muscle fibers during contraction. As a consequence, affected children show functional deficits early in childhood, eventually lose the ability to walk and can even die because of respiratory insufficiency. To date, there are no cures.
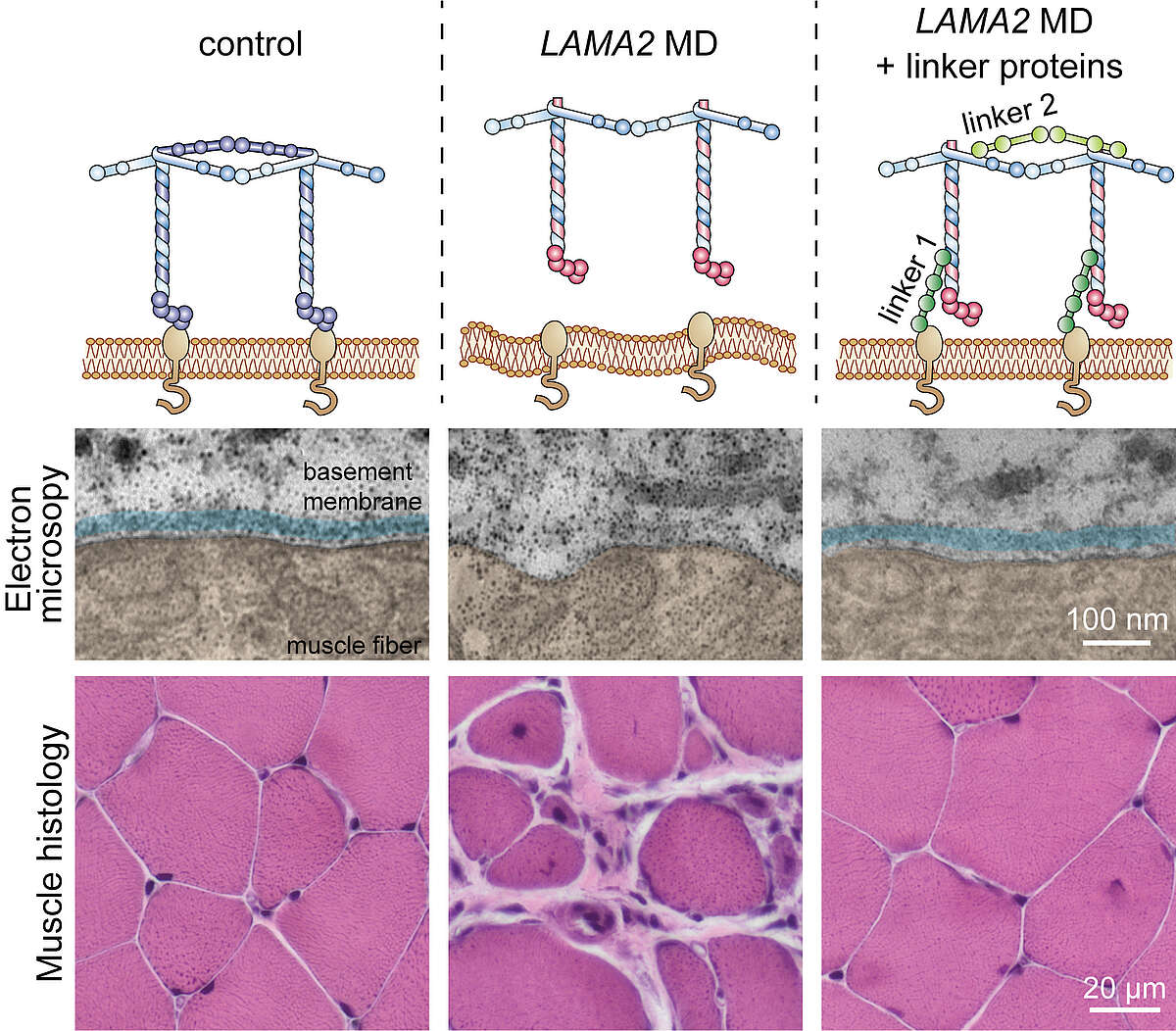
In a long-running, collaborative project we aim to rectify this for the LAMA2 MD subgroup of early-onset muscular dystrophies caused by mutations in LAMA2, the gene coding for the basement membrane protein laminin-α2. Thanks to many years of research, we have developed a potential treatment option by engineering two novel linker proteins. These linker proteins are designed to substitute for the loss of laminin-α2. Linker 1 re-connects the basement membrane to the muscle fibers, while linker 2 facilitates extracellular matrix assembly (see figure below). Using mice that carry mutations in the Lama2 gene, we first showed that expression of even a single linker is sufficient to greatly improve muscle functionality (Moll et al., 2001; McKee et al., 2017). Excitingly, simultaneous expression of both linkers restored stability of the basement membrane and normal muscle histology. The median lifespan is ~26 months in wild-type mice, but just 4 months in LAMA2 MD mice; a reduction comparable to that seen in LAMA2 patients. Expression of the two linker proteins prolonged the median lifespan in LAMA2 MD mice to almost 20 months with some reaching up to 30 months (Reinhard et al, 2017). The strength of these results has given us hope that this strategy could one day be developed into a viable treatment option for LAMA2 MD patients.

{kind=link}
Our current research focuses on addressing preclinical considerations for translation into clinical trials. Firstly, we are developing a strategy to deliver the two linkers by viral vectors and testing their efficacy to improve muscle structure and function in LAMA2 MD mice. Secondly, using tissue and temporal specific genetic models we are aiming to better understand the treatment window, target cells and capacity of the linkers to correct other clinical features of the disease. Furthermore, in an international collaboration, we are also aiming to uncover the mechanisms involved in different types of muscular dystrophies using state-of-the-art single cell proteomics and transcriptomics.
Funding:
Innosuisse
EJP RD