
LRBA deficiency is a rare and severe autoimmune disorder that was first described in 2012. This disease is caused by a genetic defect in the LRBA gene and is characterized by an impaired immune response and autoimmunity. LRBA deficiency typically manifests in early childhood and leads to a variety of health issues, such as growth and developmental delays.
The research group led by Prof. Anne Spang at the Biozentrum of the University of Basel, in collaboration with Goethe University Frankfurt, has made a significant breakthrough in understanding the cellular processes underlying this disease. In their recent study, published in “Journal of Cell Biology”, the researchers reveal that two distinct cellular recycling steps are disrupted in patients with LRBA deficiency. The findings provide new insights into the cellular mechanisms of LRBA deficiency and open up new avenues for potential therapies.
New findings on LRBA deficiency
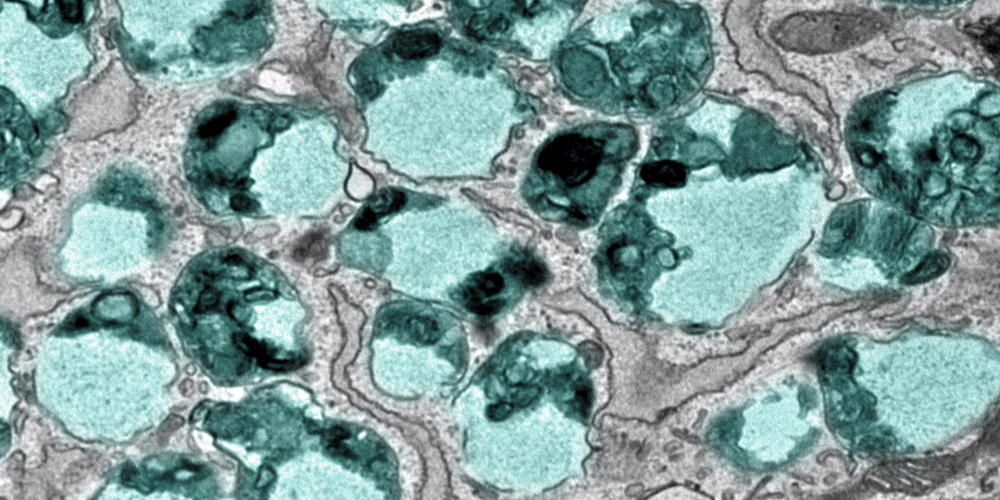
“In patients suffering from LRBA deficiency, the protein LRBA is absent in cells due to a genetic mutation,” explains first author Viktoria Szentgyörgyi. “We have now demonstrated that, unlike in healthy individuals, patient-derived cells exhibit a strong accumulation of enlarged endolysosomes.” Endolysosomes are cell organelles that play a key role in the degradation and recycling of proteins. The accumulation of enlarged endolysosomes caused by LRBA mutations leads to severe cellular dysfunctions in LRBA-deficient patients, manifesting in serious health issues such as recurrent infections and inflammatory bowel disease.
“In patient-derived cells, so-called fibroblasts, we also observed an increased secretion of endosomal and lysosomal proteins,” explains Szentgyörgyi. “Normally, endolysosomes and lysosomes degrade proteins into their basic components – amino acids – which can then be reused by the cell to synthesize new proteins”. The increased secretion of proteins by these organelles indicates a defect in the recycling process, leading to inflammation and tissue damage.
